Deep Learning for Biochemistry
Introduction to Deep Learning for Precision Medicine, Genomics, Protein Folding, Computational Chemistry. Biomedicine, Virus Identification.
Deep Learning for Precision Medicine
- Historical milestones related to precision medicine and artificial intelligence.

- Complex unresolved problems in neurodevelopmental disorders that artificial intelligence algorithms can create an impact

Deep Learning for Genomics
- Gene Editing
- Genome Sequencing
- Clinical workflows
- Consumer genomics products
- Pharmacy genomics
- Genetic screening of newborns
- Agriculture
Artificial intelligence in clinical and genomic diagnostics

Deep Learning for GWAS
-
- Code: ALS-Deeplearning
Deep Learning in Biomedicine
Course: Deep Learning in Genomics and Biomedicine
- DanQ: A Hybrid Convolutional and Recurrent Deep Neural Network for Quantifying the Function of DNA Sequences
- Basenji: Sequential regulatory activity prediction across chromosomes with convolutional neural networks
- FIDDLE: An integrative deep learning framework for functional genomic data inference
Biopython
![]()
pip3 install biopython
Genome Basics
Differences Between DNA and RNA
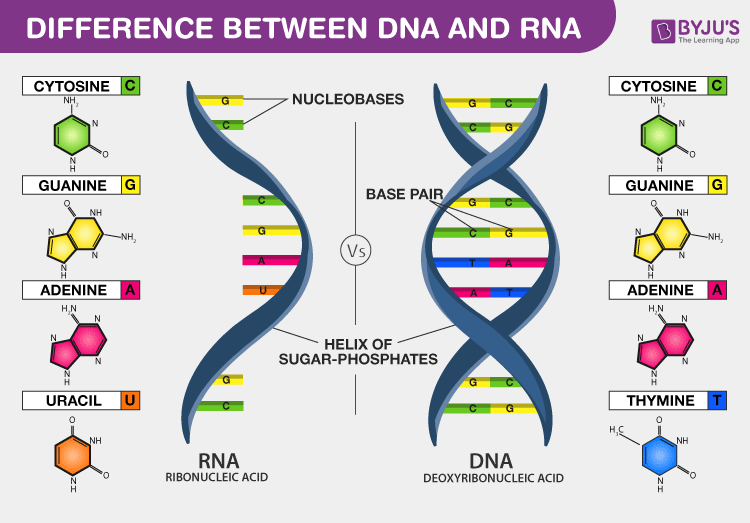 DNA vs. RNA – 5 Key Differences and Comparison
DNA vs. RNA – 5 Key Differences and Comparison
Genome, Transcriptome, Proteome, Metabolome
- Genome (基因組)
- Transcriptome (轉錄組)
- Proteome (蛋白質組)
- Metabolome (代謝組)

RNA-Seq (核糖核酸測序)
RNA-seq (核糖核酸測序)也被稱為Whole Transcriptome Shotgun Sequencing (全轉錄物組散彈槍法測序)是基於Next Generation Sequencing(第二代測序技術)的轉錄組學研究方法
Deep DNA sequence analysis
Basset
Train deep convolutional neural networks to learn highly accurate models of DNA sequence activity such as accessibility (via DNaseI-seq or ATAC-seq), protein binding (via ChIP-seq), and chromatin state.
- Basset: learning the regulatory code of the accessible genome with deep convolutional neural networks
- Deep learning models predict regulatory variants in pancreatic islets and refine type 2 diabetes association signals
ENCODE Project Common Cell Types
The Encyclopedia of DNA Elements (ENCODE) Project seeks to identify functional elements in the human genome.
- Tier 1:
- GM12878: is a lymphoblastoid cell line (淋巴母細胞系)
- K562 is an immortalized cell line (永生細胞系). It is a widely used model for cell biology, biochemistry, and erythropoiesis (紅血球細胞生成)
- H1 human embryonic stem cells
- Tier 2:
- HeLa-S3 is an immortalized cell line that was derived from a cervical cancer (宮頸癌) patient.
- HepG2 is a cell line derived from a male patient with liver carcinoma (肝癌).
- HUVEC (human umbilical vein endothelial cells) (人臍靜脈內皮細胞)
- Tier 2.5
- SK-N-SH, IMR90 (ATCC CCL-186), A549 (ATCC CCL-185), MCF7 (ATCC HTB-22), HMEC or LHCM, CD14+, CD20+, Primary heart or liver cells, Differentiated H1 cells
DeepCTCFLoop
Code: https://github.com/BioDataLearning/DeepCTCFLoop
DeepCTCFLoop is a deep learning model to predict whether a chromatin loop can be formed between a pair of convergent or tandem CTCF motifs
DeepCTCFLoop was evaluated on three different cell types GM12878, Hela and K562
- Training
python3 train.py -f Data/GM12878_pos_seq.fasta -n Data/GM12878_neg_seq.fasta -o GM12878.output
- Motif Visualization
python3 get_motifs.py -f Data/GM12878_pos_seq.fasta -n Data/GM12878_neg_seq.fasta
DARTS
Blog: 邢毅團隊利用深度學習強化RNA可變剪接分析的準確性
Paper: Deep-learning Augmented RNA-seq analysis of Transcript Splicing
 Code: https://github.com/Xinglab/DARTS
Code: https://github.com/Xinglab/DARTS
Coda
- Coda: a convolutional denoising algorithm for genome-wide ChIP-seq data
- ChIP-sequencing is a method used to analyze protein interactions with DNA.
-
ChIP-seq combines chromatin immunoprecipitation 染色質免疫沉澱 (ChIP) with massively parallel DNA sequencing to identify the binding sites of DNA-associated proteins.
- Code: https://github.com/kundajelab/coda

SNP (Single Nucleotide Polymorphism) 單核苷酸多型性
SNP(單核苷酸多型性): DNA序列中的單一鹼基對(base pair)變異,一般指變異頻率大於1%的單核苷酸變異。

- 於所有可能的DNA序列差異性中,SNP是最普遍發生的一種遺傳變異。在人體中,SNP的發生機率大約是0.1%,也就是每1200至1500個鹼基對中,就可能有一個SNP。
- 目前科學界已發現了約400萬個SNPs。平均而言,每1kb長的DNA中,就有一個SNP存在;換言之每個人的DNA序列中,每隔1kb單位長度,就至少會發生一個「單一鹼基變異」。由於SNP的發生頻率非常之高,故SNP常被當作一種基因標記(genetic marker),已用來進行研究。
DeepCpG
Paper: DeepCpG: accurate prediction of single-cell DNA methylation states using deep learning
Code: https://github.com/cangermueller/deepcpg

DeepTSS (Transcription Start Site)
Paper: Genome Functional Annotation across Species using Deep CNN
 Code: https://github.com/StudyTSS/DeepTSS
Code: https://github.com/StudyTSS/DeepTSS
Dataset: The TSS positions are collected from the reference genomes for human (hg38) and mouse (mm10) species. http://hgdownload.soe.ucsc.edu/
TSS positions over the entire human and mouse genomes data http://egg.wustl.edu/, the gene annotation is taken from RefGene
DeepFunNet
Paper: DeepFunNet: Deep Learning for Gene Functional Similarity Network Construction


- http://geneontology.org/docs/ontology-documentation/
Population Genetic Inference
Paper: The Unreasonable Effectiveness of Convolutional Neural Networks in Population Genetic Inference
 Code: https://github.com/flag0010/pop_gen_cnn
Code: https://github.com/flag0010/pop_gen_cnn
GANs for Biological Image Synthesis
Paper: GANs for Biological Image Synthesis
 Code: https://github.com/aosokin/biogans
Code: https://github.com/aosokin/biogans
Code: https://github.com/VladSkripniuk/gans
Dataset: LIN dataset
LIN dataset contains photographs of 41 proteins in fission yeast cells.
DeepGP
Genomic Selection is the breeding strategy consisting in predicting complex traits using genomic-wide genetic markers and it is standard in many animal and plant breeding schemes.
Paper: A Guide on Deep Learning for Complex Trait Genomic
Code: DLpipeine
Code: DeepGP
The DeepGP package implements Multilayer Perceptron Networks (MLP), Convolutional Neural Network (CNN), Ridge Regression and Lasso Regression to Genomic Prediction purposes.
Biochemistry Tools
PubChem Sketcher V2.4

Molview

Protein Folding
Attention Based Protein Structure Prediction
Kaggle: https://www.kaggle.com/code/basu369victor/attention-based-protein-structure-prediction
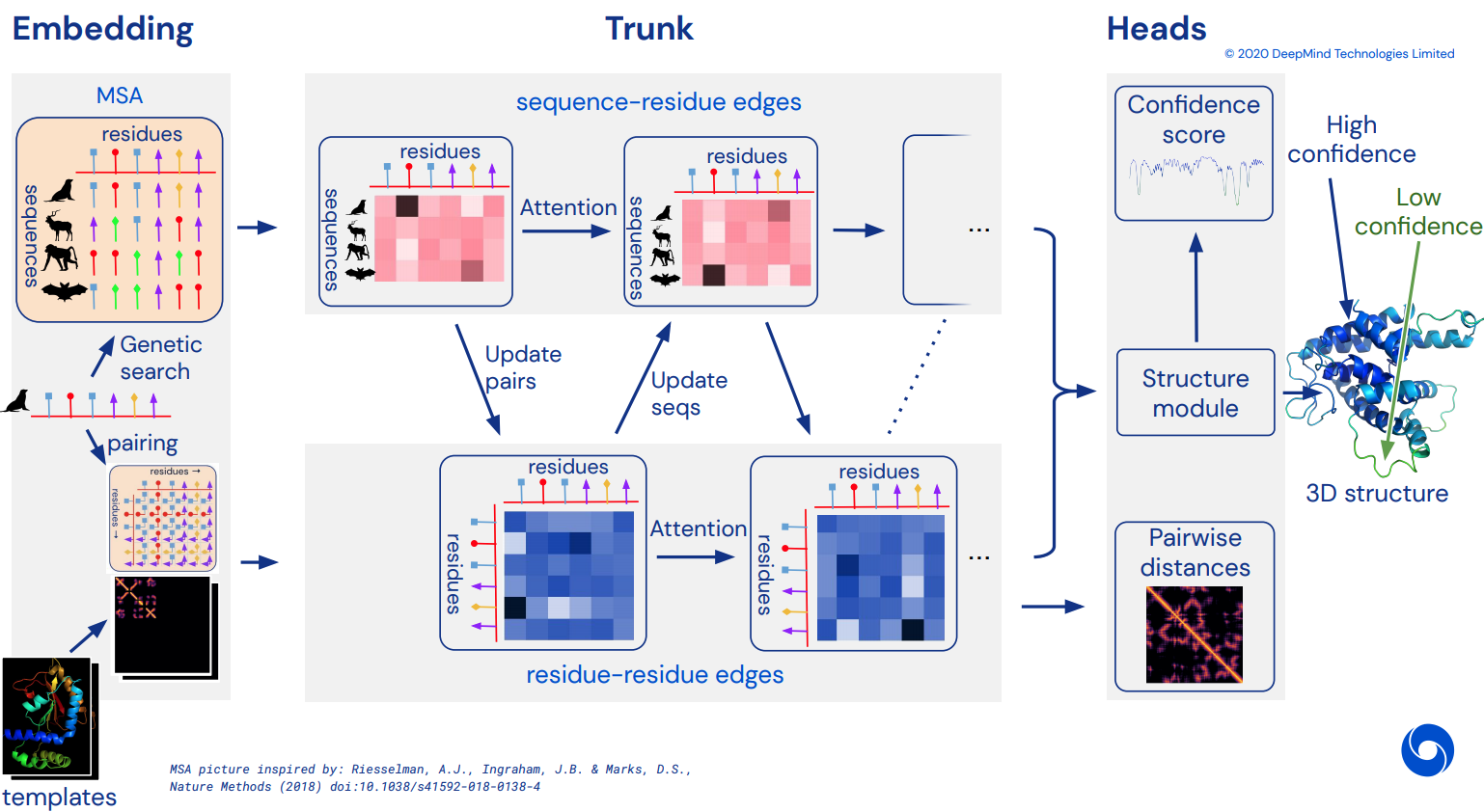
AlphaFold 2
Blog: AlphaFold reveals the structure of the protein universe
Paper: Highly accurate protein structure prediction with AlphaFold


Blog: DeepMind’s AlphaFold 2 reveal: Convolutions are out, attention is in
Code: https://github.com/deepmind/alphafold
AlphaFold Protein Structure Database
AlphaFold DB provides open access to over 200 million protein structure predictions to accelerate scientific research.
.svg)
- Q8W3K0: A potential plant disease resistance protein. Mean pLDDT 82.24.
Deep Learning for Computational Chemistry
OpenChem
![]() OpenChem is a deep learning toolkit for Computational Chemistry with PyTorch backend.
OpenChem is a deep learning toolkit for Computational Chemistry with PyTorch backend.
Code: https://github.com/Mariewelt/OpenChem
Organic Chemistry Reaction Prediction
Paper: Predicting Outcomes of Complex Organic Chemistry Reactions using Neural Sequence-to-Sequence Models
The model in version 2 is slightly based on the model discussed in Asynchronous Bidirectional Decoding for Neural Machine Translation.
Retrosynthesis Planner
Paper: Planning chemical syntheses with deep neural networks and symbolic AI
 Slides: CSC2547_learning_to_plan_chemical_synthesis.pdf
Slides: CSC2547_learning_to_plan_chemical_synthesis.pdf
Code: https://github.com/frnsys/retrosynthesis_planner
Step-wise Chemical Synthesis prediction
Code: A GGNN-GWM based step-wise framework for Chemical Synthesis Prediction


Retrosynthesis
Paper: Decomposing Retrosynthesis into Reactive Center Prediction and Molecule Generation


RetroXpert
Paper: RetroXpert: Decompose Retrosynthesis Prediction like a Chemist
 Code: https://github.com/uta-smile/RetroXpert
Code: https://github.com/uta-smile/RetroXpert
Neural Message Passing for Quantum Chemistry
Paper: Neural Message Passing for Quantum Chemistry
 A Message Passing Neural Network predicts quantum properties of an organic molecule by modeling a computationally expensive DFT calculation
A Message Passing Neural Network predicts quantum properties of an organic molecule by modeling a computationally expensive DFT calculation
Code: https://github.com/priba/nmp_qc
Biomedicine
DeepChem
Paper: Low Data Drug Discovery with One-Shot Learning
 Code: https://github.com/deepchem/deepchem
Code: https://github.com/deepchem/deepchem
druGAN
Paper: druGAN
Code: Gananath/DrugAI
Code: kumar1202/Drug-Discovery-using-GANs

MoleculeNet
Paper: MoleculeNet: A Benchmark for Molecular Machine Learning
Datasets: In most datasets, SMILES strings are used to represent input molecules
- QM7/QM7b datasets are subsets of the GDB-13 database, a database of nearly 1 billion stable and synthetically accessible organic molecules
- QM8 dataset comes from a recent study on modeling quantum mechanical calculations of electronic spectra and excited state energy of small molecules
- QM9 is a comprehensive dataset that provides geometric, energetic, electronic and thermodynamic properties for a subset of GDB-17 database
- ESOL is a small dataset consisting of water solubility data for 1128 compounds
- FreeSolv provides experimental and calculated hydration free energy of small molecules in water. Lipophilicity is an important feature of drug molecules that affects both membrane permeability and solubility. This dataset, curated from ChEMBL database, provides experimental results of octanol/water distribution coefficient (logD at pH 7.4) of 4200 compounds
- PCBA is a database consisting of biological activities of small molecules generated by high-throughput screening
- MUV group is another benchmark dataset selected from PubChem BioAssay by applying a refined nearest neighbor analysis, contains 17 challenging tasks for around 90 thousand compounds
- HIV dataset was introduced by the Drug Therapeutics Program (DTP) AIDS Antiviral Screen, which tested the ability to inhibit HIV replication for over 40,000 compounds
- Tox21 contains qualitative toxicity measurements for 8014 compounds on 12 different targets, including nuclear receptors and stress response pathways
- SIDER is a database of marketed drugs and adverse drug reactions (ADR)
- ClinTox compares drugs approved by the FDA and drugs that have failed clinical trials for toxicity reasons
TDC Datasets
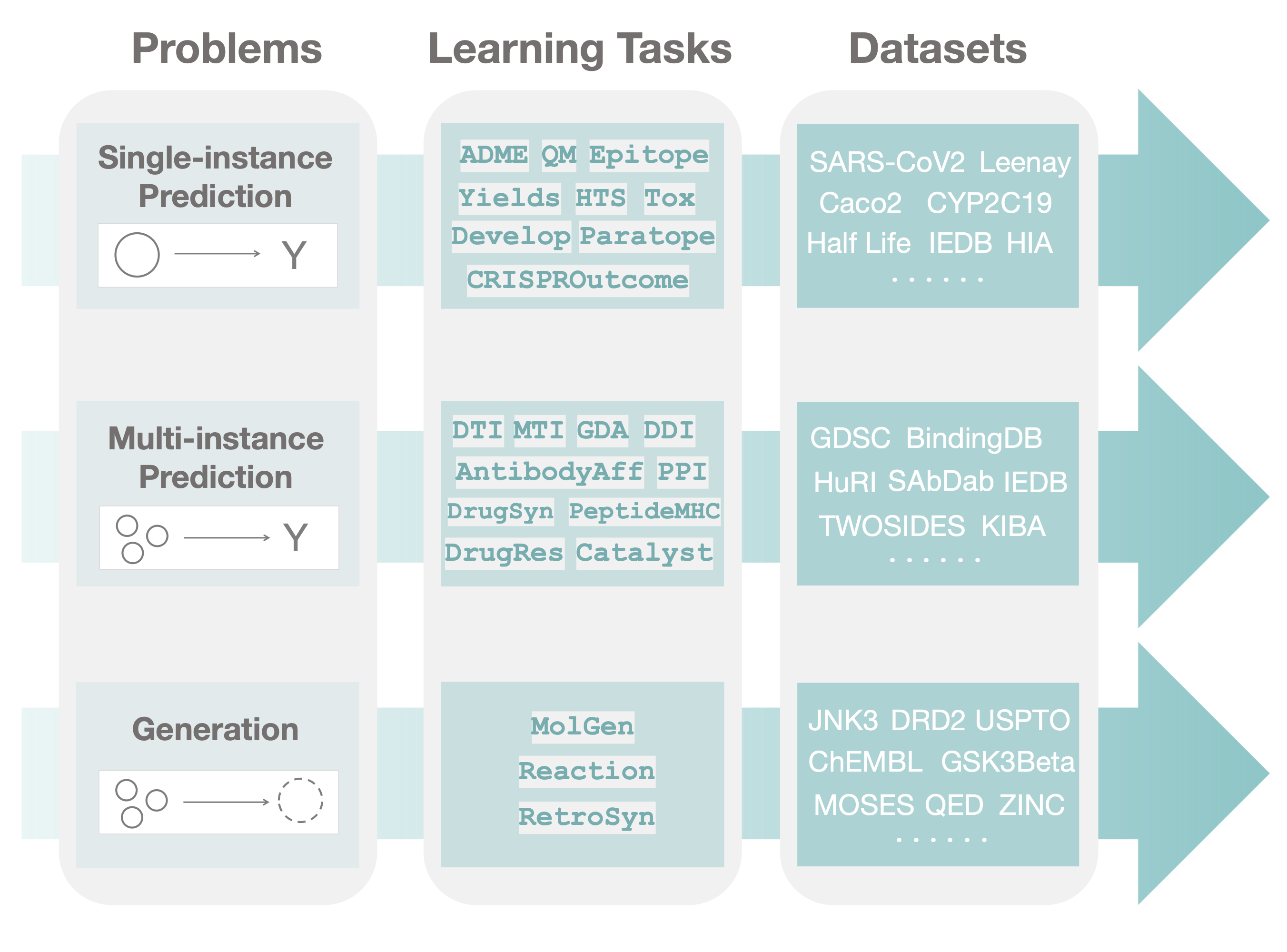
- To install PyTDC
pip3 install PyTDC - To obtain the dataset:
from tdc.Z import Y data = Y(name = ‘X’) splits = data.split() - To obtain the Caco2 dataset from ADME therapeutic task in the single-instance prediction problem:
from tdc.single_pred import ADME data = ADME(name = 'Caco2_Wang’) df = data.get_data() splits = data.get_split()
新型抗生素開發
Blog: 新型抗生素開發,機器學習立大功
- 消息傳遞神經網路
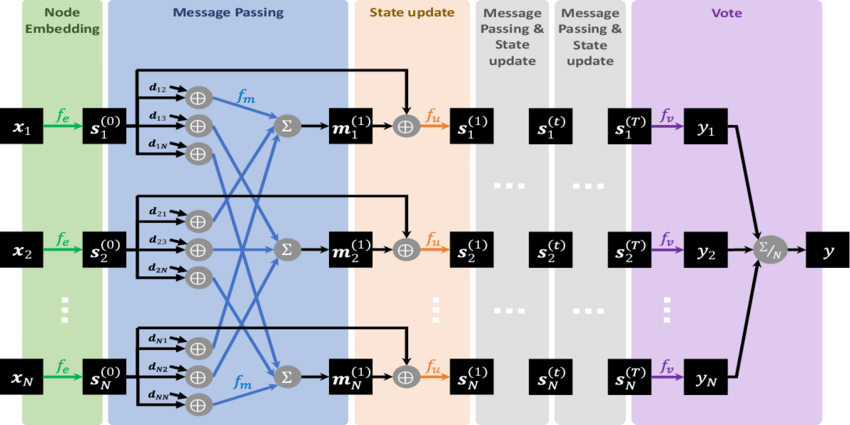 (圖片來源:M. Abdughani et al., 2019.)
(圖片來源:M. Abdughani et al., 2019.)
References:
- C. Ross, “Aided by machine learning, scientists find a novel antibiotic able to kill superbugs in mice”, STAT, 2020
- J. Gilmer et al., “Neural Message Passing for Quantum Chemistry”, arXiv.org, 2017
- G. Dahl et al., “Predicting Properties of Molecules with Machine Learning”, Google AI blog, 2017
- M. Abdughani et al., “Probing stop pair production at the LHC with graph neural networks”, Springer, 2019
Deep Learning in Proteomics
Paper: Deep Learning in Proteomics


Peptide MS/MS spectrum prediction
- pDeep3
- Reference:
- Zhou, Xie-Xuan, et al. “pDeep: predicting MS/MS spectra of peptides with deep learning.” Analytical chemistry 89.23 (2017): 12690-12697.
- Zeng, Wen-Feng, et al. “MS/MS spectrum prediction for modified peptides using pDeep2 trained by transfer learning.” Analytical chemistry 91.15 (2019): 9724-9731.
- Ching Tarn, Wen-Feng Zeng. “pDeep3: Toward More Accurate Spectrum Prediction with Fast Few-Shot Learning.” Analytical chemistry 2021.

- Reference:
- Prosit
- Code: https://github.com/kusterlab/prosit
- Webserver
- Reference:
- Gessulat, Siegfried, et al. “Prosit: proteome-wide prediction of peptide tandem mass spectra by deep learning.” Nature methods 16.6 (2019): 509-518.
- Application:
- Verbruggen, Steven, et al. “Spectral prediction features as a solution for the search space size problem in proteogenomics.” Molecular & Cellular Proteomics (2021): 100076.
- Wilhelm, M., Zolg, D.P., Graber, M. et al. Deep learning boosts sensitivity of mass spectrometry-based immunopeptidomics. Nat Commun 12, 3346 (2021).
- DeepMass
- Code: https://github.com/verilylifesciences/deepmass
- Prism is provided as a service using Google Cloud Machine Learning Engine.
- Reference:
- Tiwary, Shivani, et al. “High-quality MS/MS spectrum prediction for data-dependent and data-independent acquisition data analysis.” Nature methods 16.6 (2019): 519-525.
- Code: https://github.com/verilylifesciences/deepmass
- Predfull
- Code: https://github.com/lkytal/PredFull
- Reference:
- Liu, Kaiyuan, et al. “Full-Spectrum Prediction of Peptides Tandem Mass Spectra using Deep Neural Network.” Analytical Chemistry 92.6 (2020): 4275-4283.
- Guan et al.
- Code: https://zenodo.org/record/2652602#.X16LZZNKhT
- Reference:
- Guan, Shenheng, Michael F. Moran, and Bin Ma. “Prediction of LC-MS/MS properties of peptides from sequence by deep learning.” Molecular & Cellular Proteomics 18.10 (2019): 2099-2107.
- MS2CNN
- Code: https://github.com/changlabtw/MS2CNN
- Reference:
- Lin, Yang-Ming, Ching-Tai Chen, and Jia-Ming Chang. “MS2CNN: predicting MS/MS spectrum based on protein sequence using deep convolutional neural networks.” BMC genomics 20.9 (2019): 1-10.
- DeepDIA
- Code: https://github.com/lmsac/DeepDIA/
- Reference:
- Yang, Yi, et al. “In silico spectral libraries by deep learning facilitate data-independent acquisition proteomics.” Nature communications 11.1 (2020): 1-11.
- pDeepXL:
- Code: https://github.com/pFindStudio/pDeepXL
- Reference:
- Chen, Zhen-Lin, et al. “pDeepXL: MS/MS Spectrum Prediction for Cross-Linked Peptide Pairs by Deep Learning.” J. Proteome Res. 2021.
- Alpha-Frag:
- Code: https://github.com/YuAirLab/Alpha-Frag
- Reference:
- Jian, Song, et al. “Alpha-Frag: a deep neural network for fragment presence prediction improves peptide identification by data independent acquisition mass spectrometry.” bioRxiv. 2021.
- Prosit Transformer:
- Code: N/A
- Reference:
- Jian, Song, et al. “Prosit Transformer: A transformer for Prediction of MS2 Spectrum Intensities.” Journal of Proteome Research 2022.
- PrAI-frag
- Code: https://github.com/bertis-prai/PrAI-frag
- Webserver
- Reference:
- HyeonSeok Shin, Youngmin Park, Kyunggeun Ahn, and Sungsoo Kim “Accurate Prediction of y Ions in Beam-Type Collision-Induced Dissociation Using Deep Learning.” Analytical Chemistry May 24, 2022.
Peptide retention time prediction
- AutoRT
- Code: https://github.com/bzhanglab/AutoRT
- Reference:
- Wen, Bo, et al. “Cancer neoantigen prioritization through sensitive and reliable proteogenomics analysis.” Nature communications 11.1 (2020): 1-14.
- Application:
- Li, Kai, et al. “DeepRescore: Leveraging Deep Learning to Improve Peptide Identification in Immunopeptidomics.” Proteomics 20.21-22 (2020): 1900334.
- Rivero-Hinojosa, S., Grant, M., Panigrahi, A. et al. Proteogenomic discovery of neoantigens facilitates personalized multi-antigen targeted T cell immunotherapy for brain tumors. Nat Commun 12, 6689 (2021).
- Daisha Van Der Watt, Hannah Boekweg, Thy Truong, Amanda J Guise, Edward D Plowey, Ryan T Kelly, Samuel H Payne. “Benchmarking PSM identification tools for single cell proteomics.” bioRxiv 2021.
- Jiang W, Wen B, Li K, et al. “Deep learning-derived evaluation metrics enable effective benchmarking of computational tools for phosphopeptide identification.” Molecular & Cellular Proteomics, 2021: 100171.
- Nekrakalaya, Bhagya, et al. “Towards Phytopathogen Diagnostics? Coconut Bud Rot Pathogen Phytophthora palmivora Mycelial Proteome Analysis Informs Genome Annotation.” OMICS: A Journal of Integrative Biology (2022).
- Eric B Zheng, Li Zhao. “Systematic identification of unannotated ORFs in Drosophila reveals evolutionary heterogeneity.” bioRxiv 2022.
- Xiang H, Zhang L, Bu F, Guan X, Chen L, Zhang H, Zhao Y, Chen H, Zhang W, Li Y, Lee LJ, Mei Z, Rao Y, Gu Y, Hou Y, Mu F, Dong X. A Novel Proteogenomic Integration Strategy Expands the Breadth of Neo-Epitope Sources. Cancers. 2022; 14(12):3016.
- Prosit
- Code: https://github.com/kusterlab/prosit
- Webserver
- Reference:
- Gessulat, Siegfried, et al. “Prosit: proteome-wide prediction of peptide tandem mass spectra by deep learning.” Nature methods 16.6 (2019): 509-518.
- Application:
- Wilhelm, M., Zolg, D.P., Graber, M. et al. Deep learning boosts sensitivity of mass spectrometry-based immunopeptidomics. Nat Commun 12, 3346 (2021).
- DeepMass
- Host: https://github.com/verilylifesciences/deepmass
- DeepMass::Prism is provided as a service using Google Cloud Machine Learning Engine.
- Reference:
- Tiwary, Shivani, et al. “High-quality MS/MS spectrum prediction for data-dependent and data-independent acquisition data analysis.” Nature methods 16.6 (2019): 519-525.
- Guan et al.
- Code: https://zenodo.org/record/2652602#.X16LZZNKhT
- Reference:
- Guan, Shenheng, Michael F. Moran, and Bin Ma. “Prediction of LC-MS/MS properties of peptides from sequence by deep learning.” Molecular & Cellular Proteomics 18.10 (2019): 2099-2107.
- DeepDIA:
- Code: https://github.com/lmsac/DeepDIA
- Reference:
- Yang, Yi, et al. “In silico spectral libraries by deep learning facilitate data-independent acquisition proteomics.” Nature communications 11.1 (2020): 1-11.
- DeepRT:
- Code: https://github.com/horsepurve/DeepRTplus
- Reference:
- Ma, Chunwei, et al. “Improved peptide retention time prediction in liquid chromatography through deep learning.” Analytical chemistry 90.18 (2018): 10881-10888.
- DeepLC:
- Code: https://github.com/compomics/DeepLC
- Reference:
- Bouwmeester, R., Gabriels, R., Hulstaert, N. et al. DeepLC can predict retention times for peptides that carry as-yet unseen modifications. Nat Methods 18, 1363–1369 (2021).
- xiRT:
- Code: https://github.com/Rappsilber-Laboratory/xiRT
- Reference:
- Giese, S.H., Sinn, L.R., Wegner, F. et al. Retention time prediction using neural networks increases identifications in crosslinking mass spectrometry. Nat Commun 12, 3237 (2021).
Peptide CCS prediction
- DeepCollisionalCrossSection:
- Code: https://github.com/theislab/DeepCollisionalCrossSection
- Reference:
- Meier, F., Köhler, N.D., Brunner, AD. et al. Deep learning the collisional cross sections of the peptide universe from a million experimental values. Nat Commun 12, 1185 (2021).
Peptide detectability prediction
- CapsNet_CBAM:
- Code: yuminzhe-Prediction-of-peptide-detectability-based-on-CapsNet-and-CBAM-module
- Reference:
- Yu M, Duan Y, Li Z, Zhang Y. Prediction of Peptide Detectability Based on CapsNet and Convolutional Block Attention Module. International Journal of Molecular Sciences. 2021; 22(21):12080.
MS/MS spectrum quality prediction
- SPEQ:
- Code: https://github.com/sor8sh/SPEQ
- Reference:
- Soroosh Gholamizoj, Bin Ma. SPEQ: Quality Assessment of Peptide Tandem Mass Spectra with Deep Learning. Bioinformatics. 2022; btab874.
Peptide identification
- DeepNovo: De novo peptide sequencing
- Code: https://github.com/nh2tran/DeepNovo
- Reference:
- Tran, Ngoc Hieu, et al. “De novo peptide sequencing by deep learning.” Proceedings of the National Academy of Sciences 114.31 (2017): 8247-8252.

- Tran, Ngoc Hieu, et al. “De novo peptide sequencing by deep learning.” Proceedings of the National Academy of Sciences 114.31 (2017): 8247-8252.
- DeepNovo-DIA: De novo peptide sequencing
- Code: https://github.com/nh2tran/DeepNovo-DIA
- Reference:
- Tran, Ngoc Hieu, et al. “Deep learning enables de novo peptide sequencing from data-independent-acquisition mass spectrometry.” Nature methods 16.1 (2019): 63-66.
- SMSNet: De novo peptide sequencing
- Code: https://github.com/cmb-chula/SMSNet
- Reference:
- Karunratanakul, Korrawe, et al. “Uncovering thousands of new peptides with sequence-mask-search hybrid de novo peptide sequencing framework.” Molecular & Cellular Proteomics 18.12 (2019): 2478-2491.
- DeepRescore: Leveraging deep learning to improve peptide identification
- Code: https://github.com/bzhanglab/DeepRescore
- Reference:
- Li, Kai, et al. “DeepRescore: Leveraging Deep Learning to Improve Peptide Identification in Immunopeptidomics.” Proteomics 20.21-22 (2020): 1900334.
- PointNovo: De novo peptide sequencing
- Code: https://github.com/volpato30/PointNovo
- Reference:
- Qiao, R., Tran, N.H., Xin, L. et al. “Computationally instrument-resolution-independent de novo peptide sequencing for high-resolution devices.” Nat Mach Intell 3, 420–425 (2021).
- pValid 2: Leveraging deep learning to improve peptide identification
- Reference:
- Zhou, Wen-Jing, et al. “pValid 2: A deep learning based validation method for peptide identification in shotgun proteomics with increased discriminating power.” Journal of Proteomics (2021): 104414.

- Zhou, Wen-Jing, et al. “pValid 2: A deep learning based validation method for peptide identification in shotgun proteomics with increased discriminating power.” Journal of Proteomics (2021): 104414.
- Reference:
- Casanovo: De novo peptide sequencing
- Code: https://github.com/Noble-Lab/casanovo
- Reference:
- Melih Yilmaz, William E. Fondrie, Wout Bittremieux, Sewoong Oh, William Stafford Noble. “De novo mass spectrometry peptide sequencing with a transformer model”. bioRxiv. 2022.

- Melih Yilmaz, William E. Fondrie, Wout Bittremieux, Sewoong Oh, William Stafford Noble. “De novo mass spectrometry peptide sequencing with a transformer model”. bioRxiv. 2022.
- PepNet: De novo peptide sequencing
- Code: https://github.com/lkytal/PepNet
- Reference:
- Kaiyuan Liu, Yuzhen Ye, Haixu Tang. “PepNet: A Fully Convolutional Neural Network for De novo Peptide Sequencing”. Research Square. 2022.

- Kaiyuan Liu, Yuzhen Ye, Haixu Tang. “PepNet: A Fully Convolutional Neural Network for De novo Peptide Sequencing”. Research Square. 2022.
- DePS: De novo peptide sequencing
- Code: N/A
- Reference:
- Cheng Ge, Yi Lu, Jia Qu, Liangxu Xie, Feng Wang, Hong Zhang, Ren Kong, Shan Chang. “DePS: An improved deep learning model for de novo peptide sequencing”. arXiv. 2022.


- Cheng Ge, Yi Lu, Jia Qu, Liangxu Xie, Feng Wang, Hong Zhang, Ren Kong, Shan Chang. “DePS: An improved deep learning model for de novo peptide sequencing”. arXiv. 2022.
- DeepSCP: Utilizing deep learning to boost single-cell proteome coverage
- Code: https://github.com/XuejiangGuo/DeepSCP
- Reference:
- Bing Wang, Yue Wang, Yu Chen, Mengmeng Gao, Jie Ren, Yueshuai Guo, Chenghao Situ, Yaling Qi, Hui Zhu, Yan Li, Xuejiang Guo, DeepSCP: utilizing deep learning to boost single-cell proteome coverage. Briefings in Bioinformatics, 2022;, bbac214.

- Bing Wang, Yue Wang, Yu Chen, Mengmeng Gao, Jie Ren, Yueshuai Guo, Chenghao Situ, Yaling Qi, Hui Zhu, Yan Li, Xuejiang Guo, DeepSCP: utilizing deep learning to boost single-cell proteome coverage. Briefings in Bioinformatics, 2022;, bbac214.
Data-independent acquisition mass spectrometry
- Alpha-XIC
- Code: https://github.com/YuAirLab/Alpha-XIC
- Reference:
- Jian Song, Changbin Yu. “Alpha-XIC: a deep neural network for scoring the coelution of peak groups improves peptide identification by data-independent acquisition mass spectrometry.” Bioinformatics, btab544 (2021).
- DeepDIA:
- Code: https://github.com/lmsac/DeepDIA
- Reference:
- Yang, Yi, et al. “In silico spectral libraries by deep learning facilitate data-independent acquisition proteomics.” Nature communications 11.1 (2020): 1-11.
- DeepPhospho: impoves spectral library generation for DIA phosphoproteomics
- Code: https://github.com/weizhenFrank/DeepPhospho
- Reference:
- Lou, R., Liu, W., Li, R. et al. DeepPhospho accelerates DIA phosphoproteome profiling through in silico library generation. Nat Commun 12, 6685 (2021).
Protein post-translational modification site prediction
- DeepACE:a tool for predicting lysine acetylation sites which belong of PTM questions.
- Code: https://github.com/jiagenlee/DeepAce
- Reference:
- Zhao, Xiaowei, et al. “General and species-specific lysine acetylation site prediction using a bi-modal deep architecture.” IEEE Access 6 (2018): 63560-63569.
- Deep-PLA: for prediction of HAT/HDAC-specific acetylation
- Webserver
- Reference:
- “Deep learning based prediction of reversible HAT/HDAC-specific lysine acetylation.” Briefings in Bioinformatics (2019).
- DeepAcet: to predict the lysine acetylation sites in protein
- Code: https://github.com/Sunmile/DeepAcet
- Reference:
- “A deep learning method to more accurately recall known lysine acetylation sites.” BMC bioinformatics 20.1 (2019): 49.
- DNNAce
- Code: https://github.com/QUST-AIBBDRC/DNNAce
- Reference:
- “DNNAce: Prediction of prokaryote lysine acetylation sites through deep neural networks with multi-information fusion.” Chemometrics and Intelligent Laboratory Systems (2020): 103999.
- DeepKcr
- Code: https://github.com/linDing-group/Deep-Kcr
- Reference:
- “Deep-Kcr: Accurate detection of lysine crotonylation sites using deep learning method”, Briefings in Bioinformatics, Volume 22, Issue 4, July 2021.
- “Identification of Protein Lysine Crotonylation Sites by a Deep Learning Framework With Convolutional Neural Networks.” IEEE Access 8 (2020): 14244-14252.
- DeepGly
- Reference:
- Chen, Jingui, et al. “DeepGly: A Deep Learning Framework With Recurrent and Convolutional Neural Networks to Identify Protein Glycation Sites From Imbalanced Data.” IEEE Access 7 (2019): 142368-142378.
- Reference:
- Longetal2018
- Reference:
- Long, Haixia, et al. “A hybrid deep learning model for predicting protein hydroxylation sites.” International Journal of Molecular Sciences 19.9 (2018): 2817.
- Reference:
- MUscADEL
- Reference:
- Chen, Zhen, et al. “Large-scale comparative assessment of computational predictors for lysine post-translational modification sites.” Briefings in bioinformatics 20.6 (2019): 2267-2290.
- Reference:
- LEMP
- Reference:
- Chen, Zhen, et al. “Integration of a deep learning classifier with a random forest approach for predicting malonylation sites.” Genomics, proteomics & bioinformatics 16.6 (2018): 451-459.
- Reference:
- DeepNitro
- Reference:
- Xie, Yubin, et al. “DeepNitro: prediction of protein nitration and nitrosylation sites by deep learning.” Genomics, proteomics & bioinformatics 16.4 (2018): 294-306.
- Reference:
- MusiteDeep
- Code: https://github.com/duolinwang/MusiteDeep
- Reference:
- Wang, Duolin, et al. “MusiteDeep: a deep-learning framework for general and kinase-specific phosphorylation site prediction.” Bioinformatics 33.24 (2017): 3909-3916.
- NetPhosPan:Prediction of phosphorylation using CNNs
- Reference:
- Fenoy, Emilio, et al. “A generic deep convolutional neural network framework for prediction of receptor–ligand interactions—NetPhosPan: application to kinase phosphorylation prediction.” Bioinformatics 35.7 (2019): 1098-1107.
- Reference:
- DeepPhos
- Code: https://github.com/USTC-HIlab/DeepPhos
- Reference:
- Luo, Fenglin, et al. “DeepPhos: prediction of protein phosphorylation sites with deep learning.” Bioinformatics 35.16 (2019): 2766-2773.
- EMBER
- Code: https://github.com/gomezlab/EMBER
- Reference:
- Kirchoff, Kathryn E., and Shawn M. Gomez. “EMBER: Multi-label prediction of kinase-substrate phosphorylation events through deep learning.” BioRxiv (2020).
- DeepKinZero
- Code: https://github.com/tastanlab/DeepKinZero
- Reference:
- Deznabi, Iman, et al. “DeepKinZero: zero-shot learning for predicting kinase–phosphosite associations involving understudied kinases.” Bioinformatics 36.12 (2020): 3652-3661.
- CapsNet_PTM: CapsNet for Protein Post-translational Modification site prediction.
- Code: https://github.com/duolinwang/CapsNet_PTM
- Reference:
- Wang, Duolin, Yanchun Liang, and Dong Xu. “Capsule network for protein post-translational modification site prediction.” Bioinformatics 35.14 (2019): 2386-2394.
- GPS-Palm
- Reference:
- Ning, Wanshan, et al. “GPS-Palm: a deep learning-based graphic presentation system for the prediction of S-palmitoylation sites in proteins.” Briefings in Bioinformatics (2020).
- Reference:
- CNN-SuccSite
- Reference:
- Huang, Kai-Yao, Justin Bo-Kai Hsu, and Tzong-Yi Lee. “Characterization and Identification of Lysine Succinylation Sites based on Deep Learning Method.” Scientific reports 9.1 (2019): 1-15.

- Huang, Kai-Yao, Justin Bo-Kai Hsu, and Tzong-Yi Lee. “Characterization and Identification of Lysine Succinylation Sites based on Deep Learning Method.” Scientific reports 9.1 (2019): 1-15.
- Reference:
- DeepUbiquitylation
- Code: https://github.com/jiagenlee/deepUbiquitylation
- Reference:
- He, Fei, et al. “Large-scale prediction of protein ubiquitination sites using a multimodal deep architecture.” BMC systems biology 12.6 (2018): 109.
- DeepUbi
- Code: https://github.com/Sunmile/DeepUbi
- Reference:
- Fu, Hongli, et al. “DeepUbi: a deep learning framework for prediction of ubiquitination sites in proteins.” BMC bioinformatics 20.1 (2019): 1-10.
- Sohoko-Kcr
- Webserver
- Reference:
- Sian Soo Tng, et al. “Improved Prediction Model of Protein Lysine Crotonylation Sites Using Bidirectional Recurrent Neural Networks .” J. Proteome Res. 2021.
MHC-peptide binding prediction
- ConvMHC
- Reference:
- Han, Youngmahn, and Dongsup Kim. “Deep convolutional neural networks for pan-specific peptide-MHC class I binding prediction.” BMC bioinformatics 18.1 (2017): 585.
- Reference:
- HLA-CNN
- Code: https://github.com/uci-cbcl/HLA-bind
- Reference:
- Vang, Yeeleng S., and Xiaohui Xie. “HLA class I binding prediction via convolutional neural networks.” Bioinformatics 33.17 (2017): 2658-2665.
- DeepMHC
- Web services
- Reference:
- Hu, Jianjun, and Zhonghao Liu. “DeepMHC: Deep convolutional neural networks for high-performance peptide-MHC binding affinity prediction.” bioRxiv (2017): 239236.
- DeepSeqPan: Prediction of peptide-MHC bindings
- Code: https://github.com/pcpLiu/DeepSeqPan
- Reference:
- Liu, Zhonghao, et al. “DeepSeqPan, a novel deep convolutional neural network model for pan-specific class I HLA-peptide binding affinity prediction.” Scientific reports 9.1 (2019): 1-10.
- AI-MHC
- Webserver
- Reference:
- Sidhom, John-William, Drew Pardoll, and Alexander Baras. “AI-MHC: an allele-integrated deep learning framework for improving Class I & Class II HLA-binding predictions.” bioRxiv (2018): 318881.
- DeepSeqPanII
- Code: https://github.com/pcpLiu/DeepSeqPanII
- Reference:
- Liu, Zhonghao, et al. “DeepSeqPanII: an interpretable recurrent neural network model with attention mechanism for peptide-HLA class II binding prediction.” bioRxiv (2019): 817502.

- Liu, Zhonghao, et al. “DeepSeqPanII: an interpretable recurrent neural network model with attention mechanism for peptide-HLA class II binding prediction.” bioRxiv (2019): 817502.
- MHCSeqNet
- Code: https://github.com/cmb-chula/MHCSeqNet
- Reference:
- Phloyphisut, Poomarin, et al. “MHCSeqNet: a deep neural network model for universal MHC binding prediction.” BMC bioinformatics 20.1 (2019): 270.
- MARIA
- Reference:
- Chen, Binbin, et al. “Predicting HLA class II antigen presentation through integrated deep learning.” Nature biotechnology 37.11 (2019): 1332-1343.
- Reference:
- MHCflurry
- Code: https://github.com/openvax/mhcflurry
- Reference:
- T. O’Donnell, A. Rubinsteyn, U. Laserson. “MHCflurry 2.0: Improved pan-allele prediction of MHC I-presented peptides by incorporating antigen processing,” Cell Systems, 2020.
- O’Donnell, Timothy J., et al. “MHCflurry: open-source class I MHC binding affinity prediction.” Cell systems 7.1 (2018): 129-132.
- DeepHLApan
- Code: https://github.com/jiujiezz/deephlapan
- Reference:
- Wu, Jingcheng, et al. “DeepHLApan: a deep learning approach for neoantigen prediction considering both HLA-peptide binding and immunogenicity.” Frontiers in Immunology 10 (2019): 2559.
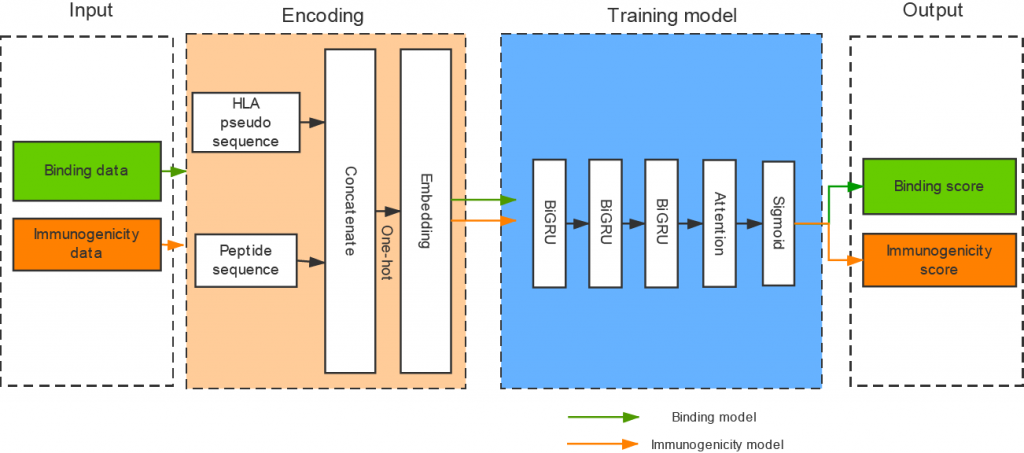
- Wu, Jingcheng, et al. “DeepHLApan: a deep learning approach for neoantigen prediction considering both HLA-peptide binding and immunogenicity.” Frontiers in Immunology 10 (2019): 2559.
- ACME
- Code: https://github.com/HYsxe/ACME
- Reference:
- Hu, Yan, et al. “ACME: pan-specific peptide–MHC class I binding prediction through attention-based deep neural networks.” Bioinformatics 35.23 (2019): 4946-4954.

- Hu, Yan, et al. “ACME: pan-specific peptide–MHC class I binding prediction through attention-based deep neural networks.” Bioinformatics 35.23 (2019): 4946-4954.
- EDGE
- Code: Supplementary data
- Reference:
- Bulik-Sullivan, Brendan, et al. “Deep learning using tumor HLA peptide mass spectrometry datasets improves neoantigen identification.” Nature biotechnology 37.1 (2019): 55-63.
- MHC-I
- Code: https://github.com/zty2009/MHC-I
- Reference:
- Zhao, Tianyi, et al. “Peptide-Major Histocompatibility Complex Class I Binding Prediction Based on Deep Learning With Novel Feature.” Frontiers in Genetics 10 (2019).
- MHCnuggets
- Code: https://github.com/KarchinLab/mhcnuggets
- Reference:
- Shao, Xiaoshan M., et al. “High-throughput prediction of MHC class i and ii neoantigens with MHCnuggets.” Cancer Immunology Research 8.3 (2020): 396-408.
- DeepNeo
- Code: DeepNeo-MHC
- Reference:
- Kim, Kwoneel, et al. “Predicting clinical benefit of immunotherapy by antigenic or functional mutations affecting tumour immunogenicity.” Nature communications 11.1 (2020): 1-11.
- DeepLigand
- Code: https://github.com/gifford-lab/DeepLigand
- Reference:
- Zeng, Haoyang, and David K. Gifford. “DeepLigand: accurate prediction of MHC class I ligands using peptide embedding.” Bioinformatics 35.14 (2019): i278-i283.
- PUFFIN
- Code: http://github.com/gifford-lab/PUFFIN
- Reference:
- Zeng, Haoyang, and David K. Gifford. “Quantification of uncertainty in peptide-MHC binding prediction improves high-affinity peptide Selection for therapeutic design.” Cell systems 9.2 (2019): 159-166.
- NeonMHC2
- Webserver
- Code: https://bitbucket.org/dharjanto-neon/neonmhc2
- Reference:
- Abelin, Jennifer G., et al. “Defining HLA-II ligand processing and binding rules with mass spectrometry enhances cancer epitope prediction.” Immunity 51.4 (2019): 766-779.
- USMPep
- Code: https://github.com/nstrodt/USMPep
- Reference:
- Vielhaben, Johanna, et al. “USMPep: universal sequence models for major histocompatibility complex binding affinity prediction.” BMC bioinformatics 21.1 (2020): 1-16.
- MHCherryPan
- Reference:
- Xie, Xuezhi, Yuanyuan Han, and Kaizhong Zhang. “MHCherryPan. a novel model to predict the binding affinity of pan-specific class I HLA-peptide.” 2019 IEEE International Conference on Bioinformatics and Biomedicine (BIBM). IEEE, 2019.
- Reference:
- DeepAttentionPan
- Code: https://github.com/jjin49/DeepAttentionPan
- Reference:
- Jin, Jing, et al. “Attention mechanism-based deep learning pan-specific model for interpretable MHC-I peptide binding prediction.” bioRxiv (2019): 830737.
Benchmarking
- Xu R, Sheng J, Bai M, et al. “A comprehensive evaluation of MS/MS spectrum prediction tools for shotgun proteomics”. Proteomics, 2020, 20(21-22): 1900345.
- Wenrong Chen, Elijah N. McCool, Liangliang Sun, Yong Zang, Xia Ning, Xiaowen Liu. “Evaluation of Machine Learning Models for Proteoform Retention and Migration Time Prediction in Top-Down Mass Spectrometry”. J. Proteome Res. (2022).
- Emily Franklin, Hannes L. Röst, “Comparing Machine Learning Architectures for the Prediction of Peptide Collisional Cross Section”. bioRxiv (2022).
Reviews about deep learning in proteomics
- Wen, B., Zeng, W.-F., Liao, Y., Shi, Z., Savage, S. R., Jiang, W., Zhang, B., “Deep Learning in Proteomics”. Proteomics 2020, 20, 1900335.
- Meyer, Jesse G. “Deep learning neural network tools for proteomics”. Cell Reports Methods (2021): 100003.
- Matthias Mann, Chanchal Kumar, Wen-Feng Zeng, Maximilian T. Strauss, Artificial intelligence for proteomics and biomarker discovery. Cell Systems 12, August 18, 2021.
- Yang, Y., Lin L., Qiao L., “Deep learning approaches for data-independent acquisition proteomics”. Expert Review of Proteomics 17 Dec 2021.
Virus Identification
ViraMiner
Paper: ViraMiner: Deep learning on raw DNA sequences for identifying viral genomes in human samples
Code: https://github.com/NeuroCSUT/ViraMiner

SAR-CoV-2
Database: SARS-CoV-2, taxid:2697049 (Nucleotide)
- SARS-CoV-2 related compounds, substances, pathways, bioassays, and more in PubChem
SARS-CoV-2 accurate identification
Paper: Accurate Identification of SARS-CoV-2 from Viral Genome Sequences using Deep Learning
Code: https://github.com/albertotonda/deep-learning-coronavirus-genome
Kaggle: rkuo2000/coronavirus-genome-identification
SARS-CoV-2 primers
Paper: Classification and specific primer design for accurate detection of SARS-CoV-2 using deep learning

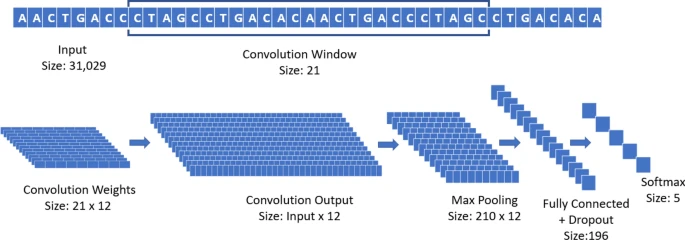 Code: https://github.com/steppenwolf0/primers-sars-cov-2
Code: https://github.com/steppenwolf0/primers-sars-cov-2
Coronavirus Typing Tool

OpenVaccine COVID-19 mRNA Vaccine Degradation Prediction
.png) Kaggle: OpenVaccine: GCN (GraphSAGE)+GRU+KFold
Kaggle: OpenVaccine: GCN (GraphSAGE)+GRU+KFold
This site was last updated October 02, 2025.